Abstract
Probiotics play a vital role in maintaining gut homeostasis, modulating immune responses, and promoting overall human health. Traditional approaches to probiotic strain development rely primarily on natural isolation and phenotypic screening, which are time-consuming and lack precision. The current study presents an in silico bioinformatics framework for the rational enhancement of probiotic strains through CRISPR-Cas9–guided design, integrated with structural bioinformatics and immunoinformatics analyses. Sequence homology and conservation were evaluated using BLAST and multiple sequence alignment to identify suitable genetic targets while minimizing off-target similarity. Structural insights were obtained from the PDB and MMDB, with PDB ID 2Z7X which was a representative immune-related protein model. Structural stability and conformational variation of hypothetical modifications were assessed using RMSD-based comparisons. Guide RNA candidates for genome editing were computationally nominated and ranked using E-CRISP and CHOPCHOP, emphasizing predicted efficiency and reduced off-target risk. To evaluate immunological safety, reverse vaccinology–based B-cell epitope prediction was performed using BepiPred, with epitope regions mapped onto three-dimensional protein structures. The integrated pipeline enables the identification of modification-tolerant regions while minimizing immunogenic potential. This purely computational strategy reduces experimental dependency, accelerates strain optimization, and provides a reproducible foundation for future probiotic engineering studies under appropriate biosafety and regulatory frameworks. This study provides a computational foundation for designing safer and more effective probiotic strains for gut health, immunomodulation, and disease prevention. The framework can support functional food development, precision microbiome therapies, vaccine-adjuvant research, and regulatory pre-screening of engineered probiotics while minimizing laboratory costs and biosafety risks.
Keywords
CRISPR-Cas9, TLR2, Engineering Probiotics, Structural Biology, Genome Editing, Immunobiotics, Structural Bioinformatic
1. Introduction
Probiotics have long been a popular part of global cultures, present in supplements and foods that boost gastrointestinal health and overall wellness. They aid in preventing diseases by maintaining a healthy balance of gut bacteria. Many fermented foods contain active microbial strains used as probiotics. Research shows that fermented foods enhance both functional and nutritional qualities by transforming substrates and generating bioactive, bioavailable end products
| [1] | Latif, A., Shehzad, A., Niazi, S., Zahid, A., Ashraf, W., Iqbal, M. W., Rehman, A., Riaz, T., Aadil, R. M., Khan, I. M., Ozogul, F., Rocha, J. M., Esatbeyoglu, T., & Korma, S. A. (2023). Probiotics: mechanism of action, health benefits and their application in food industries. Frontiers in microbiology, 14, 1216674.
https://doi.org/10.3389/fmicb.2023.1216674 |
[1]
.
Numerous studies are underway to isolate, characterize, and commercialize probiotic strains from various traditional fermented foods (TFFs). Most of these efforts mainly focus on lactic acid bacteria (LAB) and a few specialized yeasts. Probiotics provide notable benefits, such as alleviating digestive issues like diarrhea, irritable bowel syndrome, and inflammatory bowel disease, as well as enhancing overall health by boosting the immune system and supporting mental well-being. As awareness of these advantages increases, demand for probiotic-rich foods and supplements is rising, prompting further research into new strains and novel applications.
| [2] | Prajapati, K., Bisani, K., Prajapati, H. et al. Advances in probiotics research: mechanisms of action, health benefits, and limitations in applications. Syst Microbiol and Biomanuf 4, 386–406 (2024).
http://doi.org/10.1007/s43393-023-00208-w |
[2]
.
The World Health Organization (WHO) defines probiotics as live microorganisms that, when consumed in adequate amounts, confer health benefits on the host.
| [3] | Hill, C., Guarner, F., Reid, G. et al. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat Rev Gastroenterol Hepatol 11, 506–514 (2014).
https://doi.org/10.1038/nrgastro.2014.66 |
[3]
The most common probiotics include Lactobacillus, Bifidobacterium, Enterococcus, Lactococcus, and Streptococcus.
| [4] | Sarita Bhutada, Samadhan Dahikar, Hassan Md Zakir, Kovaleva Elena G. (2024). A comprehensive review of probiotics and human health-current prospective and applications. Frontiers in Microbiology, 15, Article 1487641.
https://doi.org/10.3389/fmicb.2024.1487641 |
[4]
In recent years, scientists have developed new molecular tools for gene editing, like progressing from homologous recombination to first-generation Zinc Finger Nucleases (ZFNs), second-generation Transcription Activator-Like Effector Nucleases (TALENs), and finally to the widespread use of third-generation CRISPR-Cas9 systems. Especially, ZFN and TALEN were named 'Method of the Year' by Nature Methods, while CRISPR was recognised as the 'Breakthrough of the Year' in 2015 by Science News. The significance of CRISPR-Cas9 was further confirmed when it received the 2020 Nobel Prize in Chemistry, marking a major milestone in life sciences.
This paper focuses on the use of CRISPR/Cas9 gene-editing technology to improve probiotic strains.
Emmanuelle Charpentier and Jennifer Doudna were awarded the 2020 Nobel Prize in Chemistry for their groundbreaking discovery of the CRISPR-Cas9 system, a transformative tool for targeted genome editing. Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) functions as a bacterial adaptive immune system that defends against phage infections by recognizing and cleaving foreign genetic sequences. CRISPR-associated (Cas) proteins mediate this defense. Cas9 functions as a DNA endonuclease that cleaves DNA at specific sites. Their research revealed that Cas9 is guided by a duplex formed by two RNA molecules, tracrRNA and crRNA, to achieve precise genetic targeting. They demonstrated that this duplex could be engineered into a single guide RNA (sgRNA), comprising a 20-nucleotide targeting sequence and a structure necessary for Cas9 binding. This innovation enabled the development of a simplified two-component system, allowing customizable and efficient targeting of virtually any DNA sequence, revolutionizing genetic engineering and molecular biology.
| [6] | Westermann, L., Neubauer, B., & Kotgen, M. (2021). Nobel Prize 2020 in Chemistry honors CRISPR: a tool for rewriting the code of life. Pflugers Archiv: European journal of physiology, 473(1), 1–2. https://doi.org/10.1007/s00424-020-02497-9 |
[6]
CRISPR technology depends on a thorough understanding of the relationships between gRNA sequences and gRNA performance, typically reflected in cleavage at desired and undesired sequences and in the resulting mutational profiles. Computational models have been developed to predict gRNA capabilities from massive experimental datasets, such as CRISPR screening and synthetic gRNA-target sequence libraries.
Novel approaches to deliver gut-based therapy include the CRISPR gene-editing system, which can be used to engineer the probiotic’s genome to enhance its benefits. An alternative strategy involves introducing the CRISPR system directly into the gut environment, enabling genomic alterations across multiple bacterial phyla. This method, referred to as “in situ gut microbiota modification”, allows CRISPR to function within the intestinal ecosystem itself.
| [8] | Rahmati, R., Zarimeidani, F., Ghanbari Boroujeni, M. R. et al. CRISPR-Assisted Probiotic and In Situ Engineering of Gut Microbiota: A Prospect to Modification of Metabolic Disorders. Probiotics & Antimicro. Prot. (2025).
https://doi.org/10.1007/s12602-025-10561-y |
[8]
.
TLR2, along with TLR1 or TLR6, plays a crucial role in innate immunity by detecting microbial lipoproteins and lipopeptides. When the tri-acylated lipopeptide Pam (3) CSK (4) binds, it induces the formation of an m'-shaped heterodimer between the ectodomains of TLR1 and TLR2. Conversely, the di-acylated lipopeptide Pam (2) CSK (4) does not trigger this assembly. The three lipid chains of Pam (3) CSK (4) promote receptor heterodimerization- two ester-bound chains embed into TLR2's pocket, while the amide-bound chain inserts into TLR1's hydrophobic channel. This heterodimer is stabilised by strong hydrogen bonds and hydrophobic interactions. The formation of the TLR1-TLR2 heterodimer brings their intracellular TIR domains into close proximity, facilitating dimerization and downstream signalling.
| [9] | Jin, M. S., Kim, S. E., Heo, J. Y., Lee, M. E., Kim, H. M., Paik, S. G., Lee, H., & Lee, J. O. (2007). Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell, 130(6), 1071–1082.
https://doi.org/10.1016/j.cell.2007.09.008 |
[9]
Additionally, we examine gene editing of probiotics using CRISPR-Cas9, emphasising its sustainability. Bioinformatics greatly enhances bioenergy optimisation, supports targeted conservation, and encourages future innovations through integrated data, collaboration, and creative solutions- advancing sustainability and equity. Employing bioinformatics tools reduces reliance on chemicals and laboratory work, saving potentially years and providing valuable workflow data for subsequent research.
| [10] | Sathyaseelan, C., Sankaran, D., Ravichandran, P. S., Mannu, J., Mathur, P. P. (2024). Role of Bioinformatics in Sustainable Development. In: Sobti, R. C. (eds) Role of Science and Technology for Sustainable Future. Springer, Singapore.
https://doi.org/10.1007/978-981-97-0710-2_5 |
[10]
The study utilised bioanalytical tools like PDB and MMDB for protein structure analysis and database queries, as well as UniProt for protein information retrieval. Visualisation tools such as RASMOL and PYMOL facilitated structural viewing, and BLAST was used for sequence alignment.
| [11] | Kumari, U., & Gupta, S. (2023). NGS and Sequence Analysis with Biopython for Prospective Brain Cancer Therapeutic Studies. International Journal For Science Technology And Engineering, 11(4), 3318–3329.
https://doi.org/10.22214/ijraset.2023.50885 |
| [12] | Kukreja, V., & Kumari, U. (2023). Data analysis of brain cancer with Biopython. International Journal of Innovative Science and Research Technology, 8(3), 2146–2154.
https://doi.org/10.5281/zenodo.7811128 |
| [13] | Yuan, S., Chan, H. S., & Hu, Z. (2017). Using PyMOL as a platform for computational drug design. Wiley Interdisciplinary Reviews: Computational Molecular Science, 7(2), e1298. https://doi.org/10.1002/wcms.1298 |
[11-13]
For CRISPR gene editing, E-CRISPR identified PAM sequences, while CHOPCHOP designed guide RNAS. The 2Z7X protein, involved in immune recognition, was obtained from Homo sapiens and Eptatretus burgeri- both natural producers of the protein- and was also produced through genetic engineering. Trichoplusia ni can serve as an expression system.
Recent years have witnessed significant progress in reverse vaccinology–based B-cell epitope prediction, driven by advances in machine learning, structural bioinformatics, and large-scale immunological datasets.
| [14] | Kumari, U., & Gupta, D. (2022). In silico RNA aptamer drug design and modelling. Journal of Emerging Technologies and Innovative Research, 9(4), 718–725. |
[14]
Modern reverse vaccinology frameworks increasingly integrate genome-wide antigen screening with structure-aware epitope prediction, allowing precise identification of immunogenic regions while minimizing experimental dependency. Updated B-cell epitope prediction tools now employ deep learning architectures trained on expanded epitope repositories, improving prediction accuracy compared to earlier sequence-only methods.
| [15] | Soria-Guerra, R. E., Nieto-Gomez, R., Govea-Alonso, D. O., & Rosales-Mendoza, S. (2022). An overview of bioinformatics tools for epitope prediction: Implications on vaccine development. Computational Biology and Chemistry, 97, 107645.
https://doi.org/10.1016/j.compbiolchem.2022.107645 |
[15]
.
The incorporation of protein structural data, including experimentally resolved PDB structures and high-confidence AlphaFold models, has further strengthened epitope mapping by enabling assessment of surface accessibility and conformational stability.
| [16] | Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Hassabis, D. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873), 583–589. https://doi.org/10.1038/s41586-021-03819-2 |
[16]
Recent studies emphasise combining linear epitope predictors, such as BepiPred-2.0, with structure-guided validation to discriminate between truly exposed antigenic regions and buried residues unlikely to elicit antibody responses.
.
In microbial and probiotic research, reverse vaccinology is increasingly used not only for vaccine antigen discovery but also for assessing the immunogenicity risk of engineered proteins, particularly those that interact with host immune receptors.
| [18] | Rahman, N. A., Ibrahim, M. A., & Mahmud, S. (2023). Immunoinformatics approaches for microbial protein engineering and safety assessment. Briefings in Bioinformatics, 24(5), bbad284. https://doi.org/10.1093/bib/bbad284 |
[18]
This strategy supports safer microbial design by pre-emptively identifying and avoiding immunodominant B-cell epitopes. Overall, contemporary reverse vaccinology has evolved into a robust computational framework that enhances immunological safety, precision engineering, and translational potential in microbiome and probiotic biotechnology.
2. Materials and Methods
Protein Structure and Functional Analysis Workflow.
2.1. Identification of Target Protein
The study centres on a protein sample designed to target the probiotic TOLL-like receptor 2 (TLR2) to improve gut health. This protein, with the accession code 2Z7X, is retrieved from the Protein Data Bank (PDB).
2.2. Role of the Protein Data Bank (PDB)
The PDB serves as an extensive database of experimentally verified three-dimensional structures of proteins, nucleic acids, and large molecular complexes. Every entry receives a distinct PDB ID, facilitating easy access and analysis of structural information for research purposes.
2.3. Supplementary Structural Validation Using MMDB
The Molecular Modelling Database (MMDB) was queried to obtain additional structural annotations, homology models, and domain level organization. MMDB provides curated homology models and computational structural assemblies that enhance the interpretation of protein interactions, functional sites, and structure–function relationships.
2.4. Evolutionary and Functional Characterization Using BLAST
Sequence similarity searches employed BLAST to identify conserved motifs and possible homologues. BLAST enabled rapid comparisons with reference genomes and protein databases, revealing conserved domains, evolutionary relationships, and vital functional residues. These insights were crucial for assessing off-target risks in genome editing.
2.5. Structural Visualization Using RasMol and PyMOL
The protein structure was visualized and analyzed with RasMol and Pymol. These tools allowed for detailed examination of the three-dimensional architecture, identification of functional domains, and Visualisation of potential binding sites.
2.6. Capabilities of RasMol and PyMOL
RasMol is a free molecular visualization tool that enables manipulation of protein structures, surface visualization, and domain detection. PyMOL is a more advanced molecular graphics system offering high-quality rendering, interactive editing, and molecular dynamics simulations. Both tools were crucial for visualizing the TMV protein structure as well as identifying its structural and functional features.
2.7. gRNA Design for CRISPR/Cas9 Using E CRISP and CHOPCHOP
Two complementary tools, ECRISP and CHOPCHOP, were used to design effective guide RNAs (gRNAs). ECRISP focuses on finding high-efficiency, target-specific grnas and minimises off-target effects by analyzing the genomic context and sequence uniqueness. CHOPCHOP incorporates factors like chromatin accessibility, mismatch tolerance, and PAM preferences to enhance predictions of editing efficiency and accuracy. Using both tools together ensured a trustworthy selection of the best gRNA candidates with reduced risk of unintended genomic modifications.
2.8. Integrated Methodological Framework
The workflow combines high-resolution structural data, Detailed sequence annotation, advanced visualisation tools, and accurate genome-targeting prediction algorithms. This comprehensive method enhances experimental reliability and improves understanding of the relationships between sequence, structure, and function.
2.9. Broader Impact
The integrative paradigm improves the effectiveness of CRISPR-based interventions and plays a vital role in structural bioinformatics by connecting molecular sequences, structural architectures, and biological functions within medically relevant contexts.
2.10. Reverse Vaccinology
Reverse vaccine design starts with comprehensive genomic and proteomic sequencing, which helps identify all proteins expressed by the target organism. These datasets are then analysed using in silico screening to find surface-exposed or secreted proteins, often supported by molecular docking to assess their accessibility and potential interactions with host immune receptors. After shortlisting candidate surface proteins, immunoinformatics tools predict epitopes that can provoke strong B-cell and T-cell responses. These epitopes and protein candidates are further examined for toxicity, allergenicity, stability, and similarity to host proteins to ensure safety and reduce off-target immune reactions. This process narrows down the most promising vaccine candidates. The final candidates are tested experimentally in the lab to evaluate their immunogenicity, safety, and efficacy through molecular assays, cell-based studies, and animal models, completing the reverse vaccinology process.
3. Results
3.1. Pdb Database
2Z7X | pdb_00002z7x
Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide
Classification: IMMUNE SYSTEM
Organism(s): Homo sapiens, Eptatretus burgeri, synthetic construct
Expression System: Trichoplusia ni
Mutation(s): No
3.2. Mmdb Database
Chain A, Toll-like receptor 2, Variable lymphocyte receptor B
>pdb|2Z7X|A Chain A, Toll-like receptor 2, Variable lymphocyte receptor B
SLSCDRNGICKGSSGSLNSIPSGLTEAVKSLDLSNNRITYISNSDLQRCVNLQALVLTSNGINTIEEDSFSSLGSLEHLDLSYNYLSNLSSSWFKPLSSLTFLNLLGNPYKTLGETSLFSHLTKLQILRVGNMDTFTKIQRKDFAGLTFLEELEIDASDLQSYEPKSLKSIQNVSHLILHMKQHILLLEIFVDVTSSVECLELRDTDLDTFHFSELSTGETNSLIKKFTFRNVKITDESLFQVMKLLNQISGLLELEFDDCTLNGVGNFRASDNDRVIDPGKVETLTIRRLHIPRFYLFYDLSTLYSLTERVKRITVENSKVFLVPCLLSQHLKSLEYLDLSENLMVEEYLKNSACEDAWPSLQTLILRQNHLASLEKTGETLLTLKNLTNIDISKNSFHSMPETCQWPEKMKYLNLSSTRIHSVTGCIPKTLEILDVSNNNLNLFSLNLPQLKELYISRNKLMTLPDASLLPMLLVLKISRNQLKSVPDGIFDRLTSLQKIWLHTNPWDCSCPRIDYLSRWLNKNSQKEQGSAKCSGSGKPVRSIICP
3.3. Sequcence Alignment Analysis
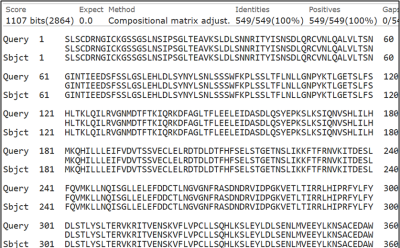
This BLAST alignment indicates a perfect match between the query and subject sequences, with 100% identity across all 549 amino acids. The alignment score is exceptionally high at 1107 bits, and the E-value is 0.0, indicating a highly significant alignment with virtually no chance of random similarity. The absence of gaps (0/549) further confirms structural and functional conservation between the sequences. Such a flawless alignment suggests that the query and subject are likely the same protein or highly conserved homologs, which is vital for gene annotation, experimental validation, and the study of evolutionary conservation in bioinformatics and molecular biology.
3.4. Rmsd Analysis
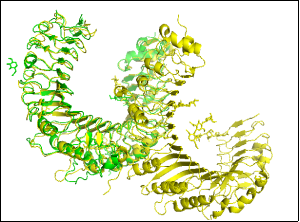
The root-mean-square analysis uses the (2z7x, 2z81) scoring matrix. It involves assigning 1,092 by 557 pairwise scores to align residues (1092 vs. 557). A total of 4,032 atoms are aligned, with an observed score RMSD of 1.190 (3400 to 3400 atoms). The root mean square score is 1.1, indicating a low RMSD. This analysis indicates a high degree of structural similarity among molecules in protein-protein docking.
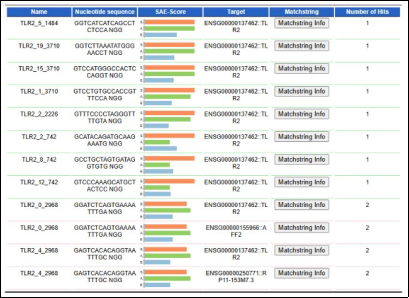
3.5. E-Crisp Analysis
In CRISPR analysis, each gRNA has a Unique Identifier that includes the transcript version and index. It also records the nucleotide sequence of the designed gRNA, including the PAM sequence (NGG), and the SAE-Score, a composite measure of gRNA efficiency, with longer bars indicating higher scores. The match string indicates the target match confidence; one hit indicates high efficiency. All candidates Grna target TLR1 with high predicted specificity and activity. Multiple transcript variants are covered (R6 and R1), SAE scoring helps to prioritizes which guides to choose, the ones with high S and A simultaneously are likely the best CRISPR candidates.
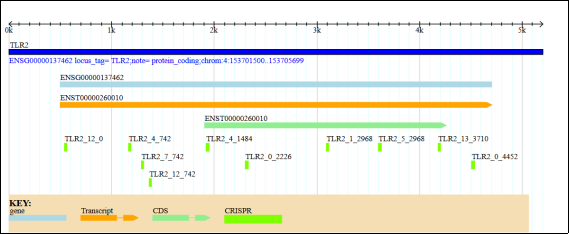
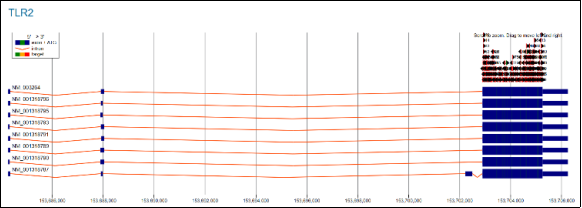
The orange arrow labelled ENST00000260010 points to a transcript variant of TLR2, while the light blue block labelled ENSG00000137462 marks the TLR2 genomic locus. The green arrow segments illustrate the coding sequence of this transcript, with each green arrow representing an exon within that sequence. In the CRISPR sites, small green bars labelled with IDS such as TLR2_12_0 and TLR2_4_742 indicate CRISPR guide (sgrna) sites, which are designed to target specific regions of TLR2.
3.6. Chopchop Analysis
Figure 5. Longitudinal visualization of TLR2 expression across the sample.
This chart shows a longitudinal analysis of TLR2 expression over two years across three experimental groups—A_1, A_2, and B_1. On the left, each horizontal line represents an individual sample from the A_1 group (e.g., A_1_HC_01 to A_1_HC_09), and the vertical bars represent expression levels. The dark blue bars indicate consistent expression intensity, while the heatmap above—comprising red and black squares—likely encodes binary or scaled molecular features, such as activation states or mutations. The timeline from January 2020 to January 2022 allows tracking of expression changes, revealing trends, stability, or shifts in TLR2 activity. The colour legend (blue for A_1, green for A_2, red for B_1) facilitates comparison of conditions, supporting both intra-group consistency checks and inter-group comparisons.
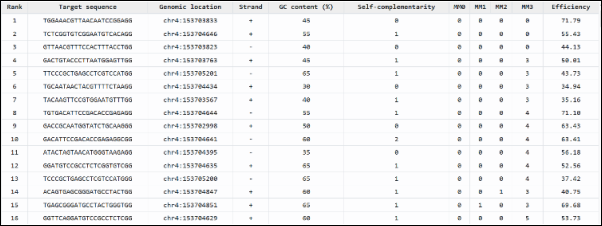
Figure 6. Summary of candidate target sequences with their rank for genome editing, including genomic location, strand orientation, and off-target potential.
This table shows a list of candidate DNA sequences for genome editing, likely for CRISPR or TALEN techniques. Each row presents a distinct target sequence with metrics indicating its suitability for precise genetic editing. The top five sequences (Ranks 1–5) are identical, measuring “TTTCTGTTGGTGCTGATGCTAGG,” and are located at the same position on chromosome 4 (chr4:23799455-23799477) on the positive strand. The GC content is moderate at 43%, suggesting balanced thermal stability. Self-complementarity is low (score of 5), reducing the chance of secondary structures. All entries have zero values for PAM, RVD, RVD2, RVD3, and off-targets, indicating high specificity and minimal risk of off-target effects. This uniformity among the top sequences demonstrates strong confidence in their targeting accuracy and their potential use in gene editing or functional genomic research.
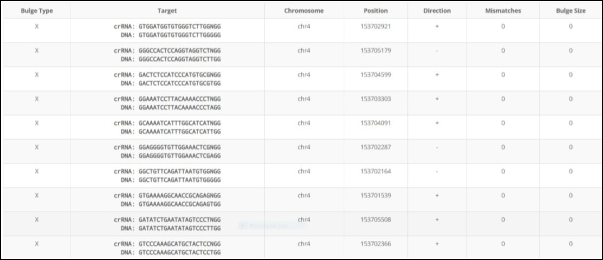
This figure illustrates that' Target CrRNA' is the guide RNA sequence used for targeting. The' Target DNA' in the genome either matches or closely resembles the crna. The' +' or' – ' signs show the direction of the DNA strand. The table reports the number of mismatches between the cRNA and the DNA target; all values are zero, indicating perfect matches or bulge-type variations. Bulges, which involve insertions or deletions (mismatches in DNA or RNA), are shown with size 0 indicating no bulge. Several loci listed in the table match the guide RNA with no mismatches or bulges, suggesting potential off-target sites. This information is essential for evaluating the specificity and safety of CRISPR gene editing, as all these sites are highly similar or identical to the guide RNA.
3.7. Reverse Vaccinology, B-Cell Epitope Prediction
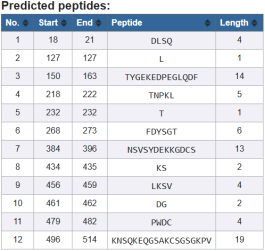
The figure displays the predicted epitope and peptide fragments from the target protein sequence. Peptides longer than 8 residues (e.g., 14, 13, 19) are particularly promising for uses like antigenicity testing, docking, and vaccine development. Shorter peptides (1–4 residues) are less likely to serve as standalone immunogenic epitopes but may still contribute to protein processing or structural motifs. This list serves as a foundation for screening, experimental validation, and prioritising peptides for further structural docking, binding affinity, and immune response studies.
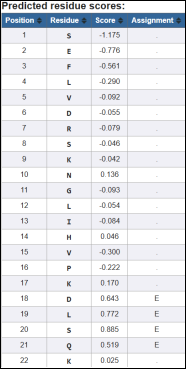
The analysis identifies a predicted epitope region between residues 18–21 (DLSQ), The consistently positive scores (>0.5) in this region and assignment as E confirm that this sequence is likely an immunogenic site. This aligns with the peptide prediction results, reinforcing the reliability of this segment as a candidate epitope for further validation (e.g., docking, vaccine design, antibody binding studies). Earlier residues (1–16) mostly have negative scores, suggesting they are unlikely to be antigenic. The region 18–21 (DLSQ) is a strong candidate epitope based on residue scoring and assignment, providing a validated target for downstream immunoinformatic or experimental studies.
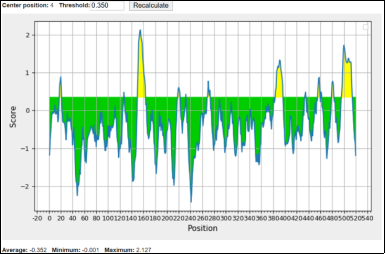
The colour-coded plot shows predicted linear B-cell epitope regions based on a score threshold of 0.35. Green areas, below the threshold, indicate residues unlikely to act as B-cell epitopes and are interpreted as non-antigenic or background regions with low probability. Conversely, yellow peaks that surpass the 0.35 threshold mark residues predicted to be linear B-cell epitopes, highlighting immunologically relevant segments for vaccine and antibody development. The protein overall features multiple yellow-highlighted regions, pointing to potential antigenic determinants. Notably, the strongest predicted epitope clusters are at positions ~140–180, ~380–400, and ~500–520, suggesting these segments may be key immunogenic sites that trigger antibody responses and are promising targets for vaccine design.
4. Discussion
Our study shows a combined computational pipeline for identifying and evaluating potential genetic targets in probiotic strains to enhance gut health functions. By merging all prediction tools, the workflow offers a step-by-step strategy for designing probiotic improvement interventions while adhering to safe, theoretical, and non-experimental parameters. The key finding of this study is the examination of a protein structure, 2Z7X, retrieved from both the PDB and MMDB databases. Structure is used as a model to analyse how structural insights can assist in selecting mutation-tolerant regions, identifying active-site proximity, and mapping epitope-bearing surface loops. Sequence analysis recognizes conserved regions and strain-specific diversity within the probiotic homologs.
| [19] | Uma Kumari, Gunika Nagpal "NEXT GENRATION SEQUENCE ANALYSIS OF AMYLOID PRECURSOR-LIKE PROTEIN 2 (APLP2) E2 DOMAIN IN ALZHEIMER’S DISEASE", International Journal of Emerging Technologies and Innovative Research, Vol. 12, Issue 1, page no. pp d72-d79, 2025. |
[19]
These results were critical for suggesting residues essential for protein function from those more amenable to engineering. Homology screening against the host and environmental microbiomes also provided early signals about potential cross-species similarities that could influence CRISPR target safety or unintended immunogenicity. Multiple sequence alignment validated these observations, elucidating distinct patterns of conserved catalytic residues and adjustable loops, some of which aligned with epitope-rich segments predicted through B-cell epitope mapping. Structural analysis using 2Z7X allowed detailed mapping of predicted epitopes, conserved residues, and candidate CRISPR-targetable regions. The representation showed that many B-cell epitopes predicted by tools such as BepiPred were concentrated in surface-exposed helices and loop regions. This aligns with known biological principles that surface accessibility influences antigenicity. By mapping both native and hypothetical mutant sequences onto the same structure, RMSD computations provided a quantitative measure of conformational deviations resulting from in silico modifications. RMSD analysis indicated that most conceptual modifications resulted in only minimal structural deviations, supporting their prioritization for further dry-lab optimization. To identify safe and efficient genome-editing target regions, CHOPCHOP and E-CRISP were used to predict guide RNAs and evaluate off-target risk. These tools generated ranked lists of candidates CRISPR target sequences based on on-target scores, GC content, and predicted off-target alignments. Integrating CHOPCHOP/E-CRISP with BLAST-based off-target screening refined the selection to potential candidates with minimal theoretical risk.
| [20] | Bandbe, T., Johri, V., Kumari, U. (2025). Structure-guided Genome-wide Association Analysis of ALK Variants with GWAS Data Using R. Computational Biology and Bioinformatics, 13(2), 72-85.
https://doi.org/10.11648/j.cbb.20251302.13 |
| [21] | Uma Kumari, Narotham BP "Integrative CRISPR-Cas9 and machine learning Approaches for Target Discovery and Therapeutic Development in Malignant Brain Tumor", International Journal of Emerging Technologies and Innovative Research (UGC and issn Approved), Vol. 12, Issue 12, page no. ppc749-c759, December-2025, Available at:
http://www.jetir.org/papers/JETIR2512289.pdf |
[20, 21]
Although this does not directly translate into operational laboratory design, it provides a strong computational rationale to inform future in vitro work conducted in accordance with appropriate biosafety protocols. In addition, the study included B-cell epitope prediction to evaluate whether engineered variants could unintentionally alter immunogenicity. Combining epitope maps with structural analysis of 2Z7X identified mutation sites with the lowest likelihood of increasing antigenic risk. This approach ensures that any theoretical modification is evaluated not only for functional impact but also for host immune compatibility, an essential consideration for probiotics intended for human use. The combination of sequence alignment, structural modeling, CRISPR target scoring, epitope mapping, and RMSD-based structural validation illustrates the strength of multi-criteria working pipelines.
| [22] | Uma Kumari, Keya Pacholee"In-silico drug discovery-based approach to treat impairments in patients of Alzheimer’s Disease", International Journal of Emerging Technologies and Innovative Research, Vol. 10, Issue 12, page no. b236-b245, December 2023, http://doi.one/10.1729/Journal.37001 |
[22]
This strategy enhances the accuracy of computational predictions by cross-verifying outcomes from independent analytical methods. The use of sample 2Z7X as a structural reference underscore how a single sample structure can substantially improve engineering decisions by providing a concrete spatial context for interpreting sequence- and epitope-based findings. Thus, the study establishes a robust computational foundation for improving probiotic strains while maintaining complete biosafety, avoiding operational editing steps, and focusing solely on theoretical and data-driven insights. These insights can guide future wet-lab experiments once appropriate ethical, regulatory, and biosafety approvals are obtained.
| [23] | Uma Kumari, Meenakshi Pradhan, Saptarshi Mukherjee, Sreyashi Chakrabarti, Ngs Analysis approach for neurodegenerative disease with Biopython, 2023, volume 10, issue 9,
http://doi.one/10.1729/Journal.36043 |
[23]
.
5. Limitations of the Study and Future Scope
This study identifies a valuable computational strategy where many limitations are found and highlights directions for future research. These experimental analyses are based on in-silico predictions. The methods are powerful, but cannot fully capture the complexity of biochemical reactions, their interactions with environmental conditions, and host interactions that determine real-world probiotic function. BLAST, CHOPCHOP, E-CRISP, and BepiPred depend mostly on available databases and predictive models that may be not include the full genetic diversity of gut microbes or strain-specific variations. Consequently, predictions may vary once experimental conditions, strain variants, or new genome annotations become available. The next limitation arises from the use of a single structural sample (PDB ID: 2Z7X) for structural mapping. Their structure effectively illustrates the pipeline’s applicability. Nevertheless, it may not accurately reflect the diversity of protein families present in probiotics. Homology modelling does not precisely replicate dynamic conformational changes or interactions with ligands or other proteins in microbial pathways. RMSD calculations are useful for estimating structural variation but are constrained by static models and do not capture long-term conformational flexibility observed in molecular dynamics simulations. Tools like CHOPCHOP and E-CRISP provide high-quality computational predictions, but their scoring algorithms are optimized using model organisms. Probiotic genomes, especially those with high GC content or unusual genomic architectures, may behave differently in real biological systems. In-silico off-target predictions may be neglect off-target sites arising from structural DNA properties or chromosomal topology. Therefore, the guide selection presented here should be viewed as a theoretical prioritization rather than a functional validation. B-cell epitope predictions also carry inherent limitations. Tools like BepiPred predict linear epitopes only and cannot predict conformational epitopes that require a complete tertiary structural context. Even with structural mapping using 2Z7X, epitope exposure may differ in solution or across strains. Additionally, immunogenicity predictions derived solely from sequence and surface mapping cannot replicate host-specific immune responses, microbiota composition, or individual variability in immune tolerance. Despite these limitations, the study creates extensive opportunities for future work. A major area of future development is the integration of multi-omics datasets—metagenomics, transcriptomics, metabolomics, and proteomics—to create a holistic model of probiotic activity. Merging In-Silico predictions with enhanced sampling via dynamic simulations could refine RMSD-based structural assessments and identify subtle structural shifts missed by static snapshots. Machine-learning-based CRISPR prediction models could enhance off-target evaluation in microbial genomes with limited experimental datasets. On the immunological side, future work could incorporate conformational epitope prediction tools, peptide-MHC binding predictions, and host-specific immunogenicity models to better model real immune interactions. Including comparative epitope analyses across multiple related strains may also help identify universally safe engineering sites that minimise unforeseen immune risks. In addition to expanding the structural dataset beyond 2Z7X to include multiple PDB/MMDB structures and integrating AlphaFold-predicted models can significantly improve the robustness of structural interpretations. This would enable larger-scale screening of engineering targets across different protein families involved in probiotic survival, metabolite production, adhesion, or pathogen antagonism. After computational predictions reach a developed stage, the next step is carefully designed laboratory experiments—conducted under appropriate biosafety approvals—that would be essential to validate predicted effects on protein function, strain competitiveness, and host interaction. The dry-lab pipeline established here ensures that such wet-lab work would be highly targeted, efficient, and grounded in systematic computational evidence.
6. Conclusions
Our current study validates an extensive computational pipeline for the justification of probiotic strains to improve gut health. Merging sequence analysis, structural modeling, epitope prediction, and CRISPR target nomination, the study provides a safe, theoretical, and data-driven framework that avoids operational genome editing steps. The use of PDB sample 2Z7X elucidates how structural findings enhance their applications by elucidating key features, preserved residues, and possible mutation-tolerant regions. Mapping predicted epitopes and computational modifications onto this structure further underscores the value of structural bioinformatics for minimizing immunogenic risks. Sequence alignment tools were used to find conserved and specific sequence motifs relevant to modification. CRISPR guide prediction through CHOPCHOP and E-CRISP, combined with off-target screening, enabled the theoretical selection of safe and effective editing targets. RMSD-based comparison between native and hypothetical modified structures provided an additional part of validation by quantifying structural deviations. B-cell epitope prediction using BepiPred added an immunological safety dimension, allowing the identification of engineering sites least likely to produce unintended antigenic changes.
| [24] | Kumari, U., Johri, V., Dhopate, S., & Jha, T. (2024). Structure based drug designing for the prediction of epitope for targeting malignant brain tumor. Journal of Emerging Technologies and Innovative Research (JETIR), 11(7). https://www.jetir.org |
[24]
Together, these components demonstrate that a multi-layer computational approach can substantially improve confidence in predicted engineering outcomes before embarking on costly or ethically demanding wet-lab procedures.
This study concludes the importance of integrating diverse computational tools to develop a comprehensive understanding of the challenges in probiotic engineering. Although experimental validation remains important for future applications, the pipeline presented here provides an efficient, reproducible, and biosafe foundation on which such work can be built.
Abbreviations
PDB | Protein Data Bank |
MMDB | Molecular Modeling Database |
BLAST | Basic Local Alignment Search Tool |
TFFs | Traditional Fermented Food |
LAB | Lactic Acid Bacteria |
ZFNs | Zinc Finger Nucleases |
TALENs | Transcription Activator-Like Effector Nucleases |
TLR | Toll-Like Receptor |
CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
gRNA | Guide RNA |
PAM | Protospacer Adjacent Motif |
Author Contributions
Uma Kumari: Conceptualization, Formal Analysis, Supervision, Writing – review & editing
Rechel Tirkey: Data Curation, Formal Analysis, Investigation, Methodology, Resources, Writing – original draft
Vipasha Rathi: Formal Analysis, Software, Writing – original draft
Funding
No external funding supports this work.
Data Availability Statement
The data is available from the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
| [1] |
Latif, A., Shehzad, A., Niazi, S., Zahid, A., Ashraf, W., Iqbal, M. W., Rehman, A., Riaz, T., Aadil, R. M., Khan, I. M., Ozogul, F., Rocha, J. M., Esatbeyoglu, T., & Korma, S. A. (2023). Probiotics: mechanism of action, health benefits and their application in food industries. Frontiers in microbiology, 14, 1216674.
https://doi.org/10.3389/fmicb.2023.1216674
|
| [2] |
Prajapati, K., Bisani, K., Prajapati, H. et al. Advances in probiotics research: mechanisms of action, health benefits, and limitations in applications. Syst Microbiol and Biomanuf 4, 386–406 (2024).
http://doi.org/10.1007/s43393-023-00208-w
|
| [3] |
Hill, C., Guarner, F., Reid, G. et al. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat Rev Gastroenterol Hepatol 11, 506–514 (2014).
https://doi.org/10.1038/nrgastro.2014.66
|
| [4] |
Sarita Bhutada, Samadhan Dahikar, Hassan Md Zakir, Kovaleva Elena G. (2024). A comprehensive review of probiotics and human health-current prospective and applications. Frontiers in Microbiology, 15, Article 1487641.
https://doi.org/10.3389/fmicb.2024.1487641
|
| [5] |
Ma, J., Lyu, Y., Liu, X. et al. Engineered probiotics. Microb Cell Fact 21, 72 (2022).
https://doi.org/10.1186/s12934-022-01799-0
|
| [6] |
Westermann, L., Neubauer, B., & Kotgen, M. (2021). Nobel Prize 2020 in Chemistry honors CRISPR: a tool for rewriting the code of life. Pflugers Archiv: European journal of physiology, 473(1), 1–2.
https://doi.org/10.1007/s00424-020-02497-9
|
| [7] |
Zhang, H., Yan, J., Lu, Z. et al. Deep sampling of gRNA in the human genome and deep-learning-informed prediction of gRNA activities. Cell Discov 9, 48 (2023).
https://doi.org/10.1038/s41421-023-00549-9
|
| [8] |
Rahmati, R., Zarimeidani, F., Ghanbari Boroujeni, M. R. et al. CRISPR-Assisted Probiotic and In Situ Engineering of Gut Microbiota: A Prospect to Modification of Metabolic Disorders. Probiotics & Antimicro. Prot. (2025).
https://doi.org/10.1007/s12602-025-10561-y
|
| [9] |
Jin, M. S., Kim, S. E., Heo, J. Y., Lee, M. E., Kim, H. M., Paik, S. G., Lee, H., & Lee, J. O. (2007). Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell, 130(6), 1071–1082.
https://doi.org/10.1016/j.cell.2007.09.008
|
| [10] |
Sathyaseelan, C., Sankaran, D., Ravichandran, P. S., Mannu, J., Mathur, P. P. (2024). Role of Bioinformatics in Sustainable Development. In: Sobti, R. C. (eds) Role of Science and Technology for Sustainable Future. Springer, Singapore.
https://doi.org/10.1007/978-981-97-0710-2_5
|
| [11] |
Kumari, U., & Gupta, S. (2023). NGS and Sequence Analysis with Biopython for Prospective Brain Cancer Therapeutic Studies. International Journal For Science Technology And Engineering, 11(4), 3318–3329.
https://doi.org/10.22214/ijraset.2023.50885
|
| [12] |
Kukreja, V., & Kumari, U. (2023). Data analysis of brain cancer with Biopython. International Journal of Innovative Science and Research Technology, 8(3), 2146–2154.
https://doi.org/10.5281/zenodo.7811128
|
| [13] |
Yuan, S., Chan, H. S., & Hu, Z. (2017). Using PyMOL as a platform for computational drug design. Wiley Interdisciplinary Reviews: Computational Molecular Science, 7(2), e1298.
https://doi.org/10.1002/wcms.1298
|
| [14] |
Kumari, U., & Gupta, D. (2022). In silico RNA aptamer drug design and modelling. Journal of Emerging Technologies and Innovative Research, 9(4), 718–725.
|
| [15] |
Soria-Guerra, R. E., Nieto-Gomez, R., Govea-Alonso, D. O., & Rosales-Mendoza, S. (2022). An overview of bioinformatics tools for epitope prediction: Implications on vaccine development. Computational Biology and Chemistry, 97, 107645.
https://doi.org/10.1016/j.compbiolchem.2022.107645
|
| [16] |
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Hassabis, D. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873), 583–589.
https://doi.org/10.1038/s41586-021-03819-2
|
| [17] |
Graham, B. S., Gilman, M. S. A., & McLellan, J. S. (2022). Structure-based vaccine antigen design. Nature Reviews Immunology, 22(12), 787–799.
https://doi.org/10.1038/s41577-022-00744-0
|
| [18] |
Rahman, N. A., Ibrahim, M. A., & Mahmud, S. (2023). Immunoinformatics approaches for microbial protein engineering and safety assessment. Briefings in Bioinformatics, 24(5), bbad284.
https://doi.org/10.1093/bib/bbad284
|
| [19] |
Uma Kumari, Gunika Nagpal "NEXT GENRATION SEQUENCE ANALYSIS OF AMYLOID PRECURSOR-LIKE PROTEIN 2 (APLP2) E2 DOMAIN IN ALZHEIMER’S DISEASE", International Journal of Emerging Technologies and Innovative Research, Vol. 12, Issue 1, page no. pp d72-d79, 2025.
|
| [20] |
Bandbe, T., Johri, V., Kumari, U. (2025). Structure-guided Genome-wide Association Analysis of ALK Variants with GWAS Data Using R. Computational Biology and Bioinformatics, 13(2), 72-85.
https://doi.org/10.11648/j.cbb.20251302.13
|
| [21] |
Uma Kumari, Narotham BP "Integrative CRISPR-Cas9 and machine learning Approaches for Target Discovery and Therapeutic Development in Malignant Brain Tumor", International Journal of Emerging Technologies and Innovative Research (UGC and issn Approved), Vol. 12, Issue 12, page no. ppc749-c759, December-2025, Available at:
http://www.jetir.org/papers/JETIR2512289.pdf
|
| [22] |
Uma Kumari, Keya Pacholee"In-silico drug discovery-based approach to treat impairments in patients of Alzheimer’s Disease", International Journal of Emerging Technologies and Innovative Research, Vol. 10, Issue 12, page no. b236-b245, December 2023,
http://doi.one/10.1729/Journal.37001
|
| [23] |
Uma Kumari, Meenakshi Pradhan, Saptarshi Mukherjee, Sreyashi Chakrabarti, Ngs Analysis approach for neurodegenerative disease with Biopython, 2023, volume 10, issue 9,
http://doi.one/10.1729/Journal.36043
|
| [24] |
Kumari, U., Johri, V., Dhopate, S., & Jha, T. (2024). Structure based drug designing for the prediction of epitope for targeting malignant brain tumor. Journal of Emerging Technologies and Innovative Research (JETIR), 11(7).
https://www.jetir.org
|
Cite This Article
-
APA Style
Kumari, U., Tirkey, R., Rathi, V. (2026). Engineering Probiotic Strains for Gut Health Enhancement Using CRISPR and Molecular Marker-assisted Technologies. Computational Biology and Bioinformatics, 14(1), 1-12. https://doi.org/10.11648/j.cbb.20261401.11
 Copy
|
Copy
|
 Download
Download
ACS Style
Kumari, U.; Tirkey, R.; Rathi, V. Engineering Probiotic Strains for Gut Health Enhancement Using CRISPR and Molecular Marker-assisted Technologies. Comput. Biol. Bioinform. 2026, 14(1), 1-12. doi: 10.11648/j.cbb.20261401.11
Copy
|
Download
AMA Style
Kumari U, Tirkey R, Rathi V. Engineering Probiotic Strains for Gut Health Enhancement Using CRISPR and Molecular Marker-assisted Technologies. Comput Biol Bioinform. 2026;14(1):1-12. doi: 10.11648/j.cbb.20261401.11
Copy
|
Download
-
@article{10.11648/j.cbb.20261401.11,
author = {Uma Kumari and Rechel Tirkey and Vipasha Rathi},
title = {Engineering Probiotic Strains for Gut Health Enhancement Using CRISPR and Molecular Marker-assisted Technologies},
journal = {Computational Biology and Bioinformatics},
volume = {14},
number = {1},
pages = {1-12},
doi = {10.11648/j.cbb.20261401.11},
url = {https://doi.org/10.11648/j.cbb.20261401.11},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.cbb.20261401.11},
abstract = {Probiotics play a vital role in maintaining gut homeostasis, modulating immune responses, and promoting overall human health. Traditional approaches to probiotic strain development rely primarily on natural isolation and phenotypic screening, which are time-consuming and lack precision. The current study presents an in silico bioinformatics framework for the rational enhancement of probiotic strains through CRISPR-Cas9–guided design, integrated with structural bioinformatics and immunoinformatics analyses. Sequence homology and conservation were evaluated using BLAST and multiple sequence alignment to identify suitable genetic targets while minimizing off-target similarity. Structural insights were obtained from the PDB and MMDB, with PDB ID 2Z7X which was a representative immune-related protein model. Structural stability and conformational variation of hypothetical modifications were assessed using RMSD-based comparisons. Guide RNA candidates for genome editing were computationally nominated and ranked using E-CRISP and CHOPCHOP, emphasizing predicted efficiency and reduced off-target risk. To evaluate immunological safety, reverse vaccinology–based B-cell epitope prediction was performed using BepiPred, with epitope regions mapped onto three-dimensional protein structures. The integrated pipeline enables the identification of modification-tolerant regions while minimizing immunogenic potential. This purely computational strategy reduces experimental dependency, accelerates strain optimization, and provides a reproducible foundation for future probiotic engineering studies under appropriate biosafety and regulatory frameworks. This study provides a computational foundation for designing safer and more effective probiotic strains for gut health, immunomodulation, and disease prevention. The framework can support functional food development, precision microbiome therapies, vaccine-adjuvant research, and regulatory pre-screening of engineered probiotics while minimizing laboratory costs and biosafety risks.},
year = {2026}
}
Copy
|
Download
-
TY - JOUR
T1 - Engineering Probiotic Strains for Gut Health Enhancement Using CRISPR and Molecular Marker-assisted Technologies
AU - Uma Kumari
AU - Rechel Tirkey
AU - Vipasha Rathi
Y1 - 2026/01/19
PY - 2026
N1 - https://doi.org/10.11648/j.cbb.20261401.11
DO - 10.11648/j.cbb.20261401.11
T2 - Computational Biology and Bioinformatics
JF - Computational Biology and Bioinformatics
JO - Computational Biology and Bioinformatics
SP - 1
EP - 12
PB - Science Publishing Group
SN - 2330-8281
UR - https://doi.org/10.11648/j.cbb.20261401.11
AB - Probiotics play a vital role in maintaining gut homeostasis, modulating immune responses, and promoting overall human health. Traditional approaches to probiotic strain development rely primarily on natural isolation and phenotypic screening, which are time-consuming and lack precision. The current study presents an in silico bioinformatics framework for the rational enhancement of probiotic strains through CRISPR-Cas9–guided design, integrated with structural bioinformatics and immunoinformatics analyses. Sequence homology and conservation were evaluated using BLAST and multiple sequence alignment to identify suitable genetic targets while minimizing off-target similarity. Structural insights were obtained from the PDB and MMDB, with PDB ID 2Z7X which was a representative immune-related protein model. Structural stability and conformational variation of hypothetical modifications were assessed using RMSD-based comparisons. Guide RNA candidates for genome editing were computationally nominated and ranked using E-CRISP and CHOPCHOP, emphasizing predicted efficiency and reduced off-target risk. To evaluate immunological safety, reverse vaccinology–based B-cell epitope prediction was performed using BepiPred, with epitope regions mapped onto three-dimensional protein structures. The integrated pipeline enables the identification of modification-tolerant regions while minimizing immunogenic potential. This purely computational strategy reduces experimental dependency, accelerates strain optimization, and provides a reproducible foundation for future probiotic engineering studies under appropriate biosafety and regulatory frameworks. This study provides a computational foundation for designing safer and more effective probiotic strains for gut health, immunomodulation, and disease prevention. The framework can support functional food development, precision microbiome therapies, vaccine-adjuvant research, and regulatory pre-screening of engineered probiotics while minimizing laboratory costs and biosafety risks.
VL - 14
IS - 1
ER -
Copy
|
Download